Pancreas endocrino
I tumori endocrini del pancreas rappresentano una quota importante fra le neoplasie ormonali digestive.
La presentazione clinica, eterogenea ed aspecifica, associata alla frequente inattività biologica, rende pressochè impossibile un'accurata determinazione della loro incidenza, bassa in lavori non recenti (1 caso/100000/anno), tanto da essere comunemente noti come tumori ad evenienza rara. Ciò contrasta con la loro alta prevalenza autoptica (1500 casi/100000/anno) che induce a riflettere sulla loro reale frequenza.
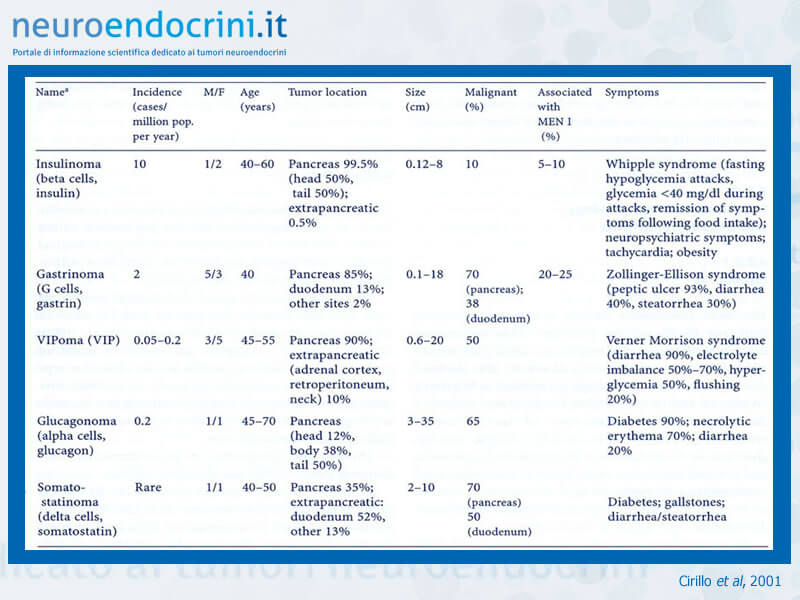
Fra i tumori insulari di più frequente osservazione, gli insulinomi rappresentano il 17%, i gastrinomi il 9%, meno del 2% i VIPomi, i glucagonomi ed i somatostatinomi. Molto più rari sono i tumori che producono ormoni ectopici ome ACTH, calcitonina, GRF e paratormone.
{kind=link}
{kind=link}
I tumori insulari sono benigni nel 70-80% dei casi. Sono per la maggior parte ben differenziati, a crescita lenta, con scarsa atipia cellulare e basso indice mitotico.
Nel 20-30% dei casi sono maligni e metastatizzano prevalentemente per via ematica al fegato.
Possono essere sporadici o con carattere di malattia familiare, diagnosticata in pazienti che appartengono a famiglie note per disordini ormonali.
Insulinoma
L'insulinoma é una neoplasia che origina dalle cellule B. La prima segnalazione si deve a von Nicholls nel 1902 ma é Whipple, qualche anno più tardi, che fornisce dell'insulinoma una descrizione clinica dettagliata, caratterizzata da crisi ipoglicemiche a digiuno associate a manifestazioni neuropsichiche, (80% dei casi), a manifestazioni cardiovascolari (10-15%) rappresentate soprattutto da tachicardia, e da sintomi gastrointestinali (4-8%) caratterizzati prevalentemente da un esasperato senso di fame (hunger pain). Si tratta del tumore insulare più frequente con una incidenza di 1/1000000/anno con un rapporto maschio/femmina di 1:2. Nel 20-30% dei casi l'anamnesi familiare é positiva per il diabete mellito; l'età più colpita é quella tra la IV e VI decade di vita.
Sono tumori di solito unici (85% dei casi) localizzati alla coda pancreatica. Il riscontro di insulinomi extrapancreatici é invece molto raro (0.5% dei casi). La malignità incide solo nel 10% dei casi e deve essere sospettata quando i sintomi ipoglicemici siano di recente comparsa. Maligni sono soprattutto i tumori di dimensioni superiori ai 2.5 cm. di diametro che presentano metastasi già al momento della diagnosi. In circa il 5-10% dei casi la neoplasia si manifesta con caratteri di familiarità .
La diagnosi di insulinoma si basa sulla valutazione dei valori glicemici e insulinemici dosati in condizioni basali oppure durante l'accesso (anche con test del digiuno protratto).
La determinazione del peptide C plasmatico risulta invece molto utile in quei pazienti che non manifestano chiari segni di ipoglicemia a digiuno.
Nella diagnostica per immagini l'ecografia ha una sensibilità variabile dallo 0 al 62%, la TC dall'11 al 73%, l'angiografia selettiva dal 35 al 67%, la risonanza magnetica nel 60% dei casi, soprattutto per la regione cefalo-pancreatica. Angiografia e TC insieme sono sensibili nel 68% dei casi .
L'ecografia endoscopica si è dimostrata in qualche caso più attendibile di altre metodiche con sensibilità talora superiori al 70% dei casi .
La chirurgia dell'insulinoma benigno può limitarsi alla semplice enucleazione del tumore dopo un attento studio imaging pre e intraoperatorio che escluda la presenza di una multifocalità; all'insulinoma maligno deve essere invece riservato un trattamento più ampio e radicale.
La resezione aggressiva di tutte le sedi metastatiche può contribuire a migliorare la qualità di vita del paziente.
Gastrinoma
Il gastrinoma origina nel pancreas da popolazioni cellulari denominate D1. Il tumore produce in modo patologico una sostanza ormonale, la gastrina, in grado di stimolare la secrezione acida dello stomaco.
La prima descrizione della neoplasia si deve a Zollinger ed Ellison nel 1955; qualche anno dopo Gregory riuscì ad isolare dalle cellule tumorali la gastrina, il peptide responsabile delle manifestazioni cliniche.
Il gastrinoma ha una incidenza approssimativa di 1/500000/anno. La localizzazione prevalente è al pancreas nel 70 % dei casi.
Le altre localizzazioni digestive sono meno frequenti (duodeno, fegato, vie biliari, digiuno, omento) ed ancora più rare le osservazioni in sedi extra-digestive, come le paratiroidi, i reni, le ovaie, lo scheletro, il polmone, la milza e il tessuto cutaneo.
L'età media, al momento della diagnosi, é intorno alla IV decade di vita; sono stati osservati anche casi in età pediatrica con una percentuale che varia dal 2 al 5%.
Il sesso maschile é il più colpito (63-71%). Il gastrinoma a sede pancreatica è maligno nel 70% dei casi; quando sintomatico, presenta una più alta incidenza metastatica.
Esso genera metastasi linfonodali con una frequenza di poco superiore rispetto a quello duodenale (39% vs 28%); la differenza é invece più netta per le metastasi epatiche (27% vs 3%) .
Il gastrinoma può essere presente come malattia familiare nel 50% dei casi. In tale caso, le paratiroidi sono interessate nell'80 % dei casi e nel 19% dei casi l'ipofisi; nell'8 % dei casi é segnalata la presenza di un insulinoma .
L'ipergastrinemia e la secondaria ipersecrezione di acido cloridrico gastrico si configurano nella sindrome di Zollinger Ellison.
Il dolore addominale (78%) e la diarrea (41%) sono le manifestazioni cliniche più importanti. Il dolore addominale è da riferire alla malattia ulcerosa nell'80% dei casi; la lesione peptica si localizza nel 75% dei casi nella porzione prossimale del duodeno e meno comunemente nello stomaco distale. Rare sono le localizzazioni al duodeno distale, al digiuno e all'esofago.
La diarrea, all'inizio intermittente, si associa a steatorrea in oltre un terzo dei pazienti; nel 7% dei casi é presente in assenza di malattia ulcerosa.
Il 7% di tutti i gastrinomi é asintomatico. In questi casi l'esordio clinico della malattia può essere diverso, come complicanza digestiva o come sindrome occupante spazio. L'emorragia digestiva é stata descritta in quasi il 46% dei casi, l'occlusione intestinale nel 20% e la perforazione nel 6% dei casi.
Il gastrinoma é presente nello 0.1-1% di tutti i pazienti affetti da malattia peptica ulcerosa: la diagnosi non é però sempre immediata (anche fino a 6 anni di latenza), in parte perchè l'epigastralgia non si presta ad essere riconosciuta come segno specifico di gastrinoma, e forse anche per l'uso indiscriminato di farmaci antisecretivi che possono mascherare le manifestazioni cliniche soprattutto nelle fasi iniziali. Tale ritardo facilita l'osservazione di metastasi al momento della diagnosi nel 50-60% dei casi.
Nel gastrinoma le concentrazioni di gastrina sierica sono sempre molto elevate (superiori a 1000 pg/ml) e permettono di formulare la diagnosi.
Per la diagnosi di sede, ecografia e TC sono sensibili nel 70% dei casi, l'angiografia nel 54% dei casi. Il sampling portale deve essere utilizzato solo quando falliscano le altre metodiche; la sua sensibilità aumenta sotto stimolo intrarterioso con secretina (test di Imamura).
L'approccio terapeutico al gastrinoma è cambiato radicalmente dopo la sintesi delle molecole antisecretive. La diagnosi di sede, sempre più accurata, ha permesso l'abbandono definitivo di interventi ampiamente demolitivi, di cui per molto tempo si è abusato, lasciando spazio ad interventi più mirati che vanno dalla semplice enucleazione del tumore pancreatico quando sicuramente unico, ben capsulato e benigno, alla più articolata chirurgia maggiore del pancreas.
La sopravvivenza a 5 anni é del 69% nei pazienti operati con criterio di radicalità, del 38% nei pazienti con malattia avanzata; a 10 anni, la sopravvivenza é rispettivamente del 62 e del 27%. Questi dati suggeriscono che una diagnosi precoce seguita da un trattamento chirurgico e farmacologico mirato potrebbero migliorare la sopravvivenza di questi pazienti o comunque ridurre il numero delle complicanze neoplastiche; una quota significativa di pazienti tende infatti a sviluppare localizzazioni anche allo scheletro, compromettendo ulteriormente la prognosi della malattia.
VIPoma
Nel 1958 Verner e Morrison descrissero in due pazienti una sindrome caratterizzata da diarrea, ipokaliemia, ipocloridria e conseguente acidosi metabolica. Fu coniato per questa sindrome l'acronimo WDHA (Watery Diarrhoea, Hypokaliemia, Achlorydria) modificato nel 1974 in WDHH (Watery Diarrhoea, Hypokaliemia, Hypochlorydria).
La sindrome é stata anche definita "colera pancreatico": tale definizione diventa però inappropriata quando la malattia si presenta in sedi extrapancreatiche.
Il riscontro di elevate concentrazioni plasmatiche di VIP (Polipeptide Vasoattivo Intestinale) ha suggerito anche il termine di VIPoma.
In letteratura sono stati riportati oltre 250 casi di VIPoma. L'età media al momento della diagnosi é di 50 anni. Prevale il sesso femminile.
Si tratta di un tumore a sede unica nell'80% dei casi, con un diametro variabile da 1 a 7 cm (20 cm in un caso segnalato).
L'80% dei VIPomi è localizzato al pancreas ed è maligno nella metà dei casi. Nel 10% dei casi è a sede extrapancreatica, localizzato nelle ghiandole surrenali, nel retroperitoneo e nel collo. Nell'infanzia, gli alti livelli sierici di VIP sono di solito legati alla presenza di un neuroblastoma.
La sindrome da VIPoma é caratterizzata da diarrea cospicua (1-6 lt / die), acquosa e ingravescente. I disturbi dell'equilibrio idro-elettrolitico secondari alla diarrea possono portare a letargia, debolezza muscolare, disturbi della conduzione cardiaca, calo ponderale, addominalgia, dispepsia. Nel 20% dei casi é presente flushing e più raramente necrosi tubulare renale acuta.
Nel 75% dei pazienti si riscontra ipercalcemia accompagnata da ipofosfatemia della quale può essere responsabile un iperparatiroidismo associato. Anche la stessa secrezione ormonale neoplastica può essere in grado di indurre ipercalcemia.
La metà dei pazienti é affetta da iperglicemia per ridotta tolleranza al glucosio; nel 40% dei casi é presente ipo-acloridria. Episodi di flushing sono segnalati nel 20% dei casi.
Nelle fasi iniziali, la sindrome di Verner-Morrison può essere confusa con forme diarroiche di altra origine, come quella batterica, virale, parassitaria, oppure con forme di tipo ulcerativo (colite ulcerosa, morbo di Crohn). La diagnosi differenziale deve essere posta anche con le altre neoplasie neuroendocrine diarrogene.
Nei pazienti affetti da VIPoma le concentrazioni plasmatiche di VIP (presente in diverse forme molecolari) sono molto elevate. Spesso risultano elevate anche le concentrazioni sieriche di altre sostanze diarrogene quali la secretina, il polipeptide pancreatico (PP), il polipeptide inibitorio gastrico (GIP), la prostaglandina E2 e il PHI.
L'imaging diagnostico è facilitato dalle dimensioni del tumore. In assenza di lesioni pancreatiche documentabili diviene obbligatoria l'esplorazione delle ghiandole surrenali e del plesso simpatico allo scopo di escludere una localizzazione extrapancreatica.
Glucagonoma
La secrezione di glucagone dalle cellule A delle insule pancreatiche gioca un ruolo importante nel controllo del metabolismo glucidico. Una secrezione inappropriata di questo ormone genera una sindrome, descritta per la prima volta da Becker nel 1942, caratterizzata da manifestazioni patologiche della pelle quali l'eritema necrolitico migrante di Wilkinson, la stomatite angolare, la cheilite, la glossite atrofica e, più raramente, la porpora. La patogenesi di queste lesioni cutanee é probabilmente multifattoriale. La ridotta concentrazione aminoacidica nel sangue (alanina e glutamina) potrebbe essere una delle cause delle manifestazioni cutanee, tanto da poter escludere una stretta relazione tra queste e le alte concentrazioni plasmatiche di glucagone.
Oltre alle lesioni cutanee, altri segni della malattia sono l'importante calo ponderale, l'anemia normocromica normocitica, la ridotta concentrazione plasmatica di aminoacidi (alanina e glutamina), di acidi grassi essenziali e di zinco conseguenti all'attività catabolica esercitata dal glucagone. In tutti i pazienti si riscontra diabete mellito, talvolta resistente al trattamento insulinico. La diarrea é presente nel 20% dei pazienti della quale può essere responsabile la ipersecrezione di altri enterormoni. Negli altri casi é invece presente costipazione dell'alvo fino al manifestarsi di episodi sub-occlusivi: il glucagone é infatti un potente inibitore della motilità intestinale.
Sono state segnalate anche alterazioni neurologiche delle quali potrebbe essere responsabile un fattore circolante capace di indurre la comparsa di una encefalomielite.
Fra le altre manifestazioni di non chiara patogenesi, la flebotrombosi e l'embolia polmonare risultano così frequenti da giustificare indicato il trattamento profilattico.
In letteratura sono riportati oltre 200 casi di glucagonoma, ma per l'aspecificità dei sintomi si ritiene che la malattia possa avere una incidenza più alta (0.2/1000000/anno). L'età media, al momento della diagnosi, é intorno ai 65 anni (19-84), il 75% dei casi fra i 45 e i 70. Non si osserva prevalenza nella distribuzione dei sessi.
Raro è il glucagonoma osservato nell'ambito di forme familiari nelle quali possono coesistere elevate secrezioni di altri peptidi come insulina, gastrina, somatostatina, VIP e PP.
La presenza di un glucagonoma deve essere sempre sospettata in ogni paziente diabetico che presenti il caratteristico rash cutaneo. La diagnosi trova conferma negli elevati valori plasmatici di glucagone.
Il tumore, a causa dei sintomi aspecifici, viene di solito diagnosticato in ritardo quando già metastatico; nel 60-70% dei casi ha infatti caratteri di malignità. Le metastasi, in prevalenza epatiche, possono essere riscontrate anche a livello vertebrale, surrenale e linfonodale. Nel 50-77% dei casi il tumore é localizzato alla coda del pancreas dove può raggiungere i 5-10 cm di diametro. Prevalgono le forme funzionanti.
Allo stato attuale delle conoscenze, quello chirurgico é considerato l'unico trattamento efficace con risultati soddisfacenti in meno di un terzo dei casi. La chemioterapia garantisce un buon numero di risposte senza però prolungare la sopravvivenza; sono segnalati casi di remissione parziale dopo trattamento con analoghi della somatostatina ed interferone. La recidiva metastatica é molto frequente.
Somatostatinoma
La somatostatina é un ormone identificato per la prima volta nelle cellule pituitarie come regolatore metabolico dell'ormone della crescita. È prodotto dalle cellule D che sono state osservate nel pancreas ed in tutto l'apparato gastroenterico. La prima descrizione di un tumore a prevalente secrezione di somatostatina si deve a Larsson, Ganda e Kovacs nel 1977.
La neoplasia può essere biologicamente attiva o asintomatica. In quest'ultimo caso, la diagnosi è il più delle volte secondaria ad ostruzione biliare, data la predilezione della malattia per la regione ampollare. Quando sintomatica, la neoplasia é caratterizzata da una sindrome con manifestazioni cliniche estremamente variabili che riflettono tutte le azioni farmacologiche della somatostatina.
È presente un diabete acquisito, anche se di lieve entità, per l'effetto inibitorio della somatostatina sia sull'insulina che sul glucagone; la stasi biliare, secondaria alla inibizione della colecisti, é responsabile della calcolosi (81%); l'alterata motilità biliare ed intestinale in associazione alla ridotta secrezione pancreatica sono responsabili della diarrea e della steatorrea (78%). Possono essere presenti anche anemia ipocromica (55%), calo ponderale (43%) e ipo-acloridria (30%).
La classica triade diabete/colelitiasi/diarrea-steatorrea può però essere arricchita da manifestazioni cliniche diverse, secondarie alla produzione di altri enterormoni.
La neoplasia incide tra la IV e V decade di vita, senza particolare predilezione per il sesso. Più frequente nel duodeno (52%) che nel pancreas (35%), dove la neoplasia é di solito attiva, il somatostatinoma é caratterizzato da un grado di malignità considerevole, diverso a seconda della sede di localizzazione primitiva: 70% per il pancreas, 50% per il duodeno. Nei rari casi di somatostatinoma extra-digestivo la malignità é la regola.
Nella maggior parte dei casi la diagnosi di somatostatinoma é retrospettiva a causa della aspecificità delle manifestazioni cliniche. In altri casi il tumore viene riconosciuto per caso nel corso di una laparotomia per colelitiasi oppure in seguito ad altre indagini rese obbligatorie dalle complicanze della neoplasia (emorragia, colica addominale, ittero). In questi casi il somatostatinoma si presenta di cospicue dimensioni (>5 cm di diametro) e metastatico nella maggior parte dei casi (74%). La diagnosi definitiva é posta quando la concentrazione plasmatica di somatostatina supera le 100 pmol/lt.
Nel 75% dei casi é presente la secrezione di altri ormoni (calcitonina, PP, glucagone, ACTH) che possono mutare il quadro clinico. La sopravvivenza, indipendentemente dal trattamento, è presente nel 48% dei casi ad un anno di distanza dalla diagnosi.
Tumore secernente polipeptide pancreatico
Concentrazioni plasmatiche di polipeptide pancreatico (PP, detto anche GIF: Growth Inhibiting Factor; oppure hPP: human Pancreatic Polipeptide) vengono riscontrate con relativa frequenza in pazienti affetti da tumore insulare (75% dei pazienti con VIPoma, 25% con gastrinoma), in pazienti con neoplasie endocrine multiple e, talora, in pazienti con tumori intestinali o bronchiali.
La sede di insorgenza più frequente del PPoma è la testa del pancreas. Le dimensioni del tumore sono cospicue (>5 cm di diametro), tali da determinare una sindrome occupante spazio. La localizzazione del PPoma è possibile con le metodiche radiologiche tradizionali.
Il trattamento chirurgico è quello di scelta associato, quando possibile, ad una generosa citoriduzione delle metastasi.
Nei casi di PPoma finora descritti in letteratura, i segni e i sintomi riportati sono: diarrea acquosa, lieve ipergastrinemia, ulcera peptica (che permette la diagnosi differenziale col VIPoma), ipersecretinemia, elevati valori di secrezione acida basale e di PP plasmatico, ipopotassiemia. È stato pure descritto un rash cutaneo simile a quello caratteristico del glucagonoma, che si risolve con l'asportazione della neoplasia.
Tumore secernente neurotensina
Alcuni tumori pancreatici, erroneamente classificati come VIPomi, secernono un neuropeptide, la neurotensina, che ha in comune con questi diverse azioni biologiche. La neoplasia ha origine dalle cellule N; ne sono stati descritti meno di 40 casi prevalentemente localizzati al pancreas.
Quando biologicamente attivo il neurotensinoma presenta manifestazioni cliniche quasi del tutto simili a quelle descritte per i pazienti affetti da VIPoma: diarrea, ipotensione, tachicardia, cianosi, reflusso esofageo.
Tuttavia, i tumori secernenti neurotensina producono anche altri enterormoni (VIP con maggiore frequenza) così che la sintomatologia può essere estremamente variabile. È stato anche segnalato un caso di neurotensinoma secernente contemporaneamente calcitonina ed encefalina.
Carcinoide pancreatico
Dal 1838 (anno della prima descrizione di un carcinoide intestinale ad opera di Merling) al 1998 sono stati osservati 43 casi di carcinoide pancreatico, pari allo 0.5% di tutti i tumori neuroendocrini gastroenteropancreatici.
Si tratta di una varietà molto rara che predilige la testa e la coda pancreatica rispettivamente, mentre negli altri casi (29%) é a localizzazione multipla.
I pazienti sono di sesso maschile, nella V decade di vita.
La sindrome da carcinoide è presente nel 64% dei casi. Nell'88% si tratta di neoplasie maligne giunte a diagnosi con malattia già metastatica nella quasi totalità dei casi.
Le possibilità terapeutiche sono molto povere, condizionate anche da una diagnosi ritardata e da una bassa incidenza di resecabilità, con dati incerti di sopravvivenza dopo resezione.
Altri tumori meno frequenti
Sono state riportate in letteratura sporadiche segnalazioni di tumori insulari secernenti altri enterormoni (CCK, GIP, enteroglucagone) o con secrezione ectopica di ormoni extra-digestivi o di releasing factors (PTH, ACTH, GRF).
Le manifestazioni cliniche sono direttamente legate all'azione biologica dell'ormone secreto. In un caso di GIPoma riportato, la manifestazione clinica più importante é la diarrea acquosa responsabile dello squilibrio idro-elettrolitico. Di questo tumore é stata però messa in dubbio la diagnosi in quanto il GIP ha reattività crociata con il VIP.
Anche nel CCKoma la diarrea acquosa é il segno più importante con grave ipercloridria normogastrinemica ed aumentati livelli sierici di colecistochinina.
Nelle neoplasie a prevalente secrezione di enteroglucagone (cellule L) sono state riscontrate iperglicemia, iperenteroglucagonemia, ipoacidità gastrica, rallentamento della motilità intestinale, malassorbimento ed ipertrofia villosa della mucosa enterica.
Tale neoplasia, detta anche glicentinoma, è stata fino ad ora segnalata in meno di 40 casi.
È stato segnalato un caso di tumore pancreatico secernente amilina (IAPPoma, Islet Amyloid Polypeptide Producing) caratterizzato da flushing, diarrea e iperglicemia, sensibile alla terapia con interferone; un caso di motilinoma, biologicamente inattivo, é risultato invece refrattario ad ogni trattamento.
Un caso di bombesinoma (GRPoma) biologicamente attivo, localizzato nella regione cefalo-pancreatica, presentava diabete, flushing e gastrite emorragica; quest'ultima è in rapporto con un eccesso plasmatico di GRP (Gastrin Releasing Peptide), analogo della bombesina e potente stimolo per la secrezione di gastrina. La relazione tra diabete, flushing e GRP non è invece chiara . Nel caso descritto i livelli di GRP plasmatico erano 20 volte superiori rispetto al soggetto sano.
Sono stati descritti 80 casi di paraganglioma gangliocitico (GPG) di cui solo uno biologicamente attivo a sede cefalo-pancreatica, caratterizzati da dolore addominale, steatorrea e vomito. Di solito benigni, metastatici in 7 casi (8.7%). L'ormone più frequentemente prodotto è la somatostatina.
Undici sono i casi di GRFoma pancreatico, producenti il releasing factor per l'ormone della crescita. Sono associati ad acromegalia, diabete e ipersecrezione di GH. L'età media dei pazienti è di 40 anni (range 15-60) con una frequenza 3 volte maggiore nel sesso femminile rispetto all'acromegalia classica. Nel 40% dei casi, il GRFoma è associato alla secrezione di altri peptidi nell'ambito di una MEN 1; nel 70% dei casi è presente una iperprolattinemia secondaria a stimolo diretto da parte di GRF. In un caso segnalato, il trattamento con analogo della somatostatina ha mostrato risposte obiettive incoraggianti.
Tumori biologicamente inattivi
Presenti nel 50% dei casi, sono clinicamente silenti. La maggior parte di questi tumori viene diagnosticata quando determina una sindrome occupante spazio. Nel 70% dei casi essi si localizzano a livello cefalo-pancreatico. Sono tumori maligni nella quasi totalità dei casi, metastatici al momento della diagnosi nel 50-70% dei casi, interessando prevalentemente fegato, linfonodi regionali, peritoneo, polmone, rene e scheletro. La resezione a scopo curativo è possibile solo nel 20% dei casi.
{kind=link}